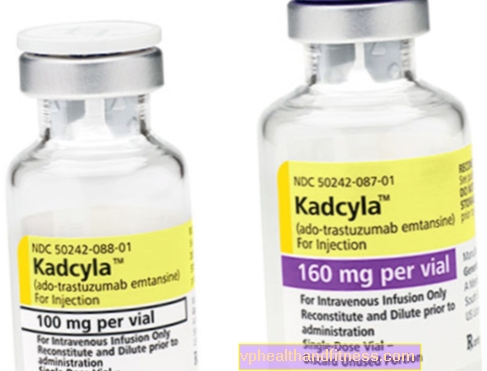
1 injekční lahvička obsahuje 100 mg (160 mg) prášku pro rekonstituci 5 ml (8 ml) roztoku koncentrátu trastuzumab emtansinu v koncentraci 20 mg / ml pro infuzi po rekonstituci.
| název | Obsah balení | Léčivá látka | Cena 100% | Naposledy změněno |
| Kadcyla | 1 lahvička, prášek pro přípravu konečné řešení do inf. | Trastuzumab emtansin | 2019-04-05 |
Akce
Protinádorová droga. Trastuzumab emtansin je konjugát protilátka-léčivo, který obsahuje trastuzumab, humanizovanou monoklonální protilátku anti-HER2 IgG1, kovalentně navázanou na DM1 (derivát maytansinu, inhibitor mikrotubulů) prostřednictvím stabilního MCC thioetherového spojení (4- cyklohexan-1-karboxylát) . Emtansin je komplex MCC-DM1. Spojení DM1 s trastuzumabem způsobuje selektivní působení cytotoxických léků proti HER2 nadměrně exprimujícím nádorovým buňkám a zvyšuje tak intracelulární koncentraci DM1 přímo v nádorových buňkách. Po navázání na HER2 je trastuzumab emtansin receptorem zprostředkovaná internalizace následovaná lysozomální degradací a uvolněním katabolitů obsahujících DM1 (primárně lysin-MCC-DM1). Mechanismus účinku trastuzumabu emtansinu je zprostředkován aktivitou trastuzumabu a DM1. Trastuzumab emtansin se podobně jako trastuzumab váže na doménu IV extracelulární domény (ECD) receptoru, stejně jako na receptory Fcy a komplementuje C1q. Kromě toho inhibuje aktivitu ECD domény receptoru HER2, inhibuje signalizaci dráhy fosfatidylinositol-3-kinázy (PI3-K) a zprostředkovává na protilátkách závislou buněčnou cytotoxicitu (ADCC) v lidských buňkách rakoviny prsu nadměrně exprimujících HER2. DM1 se váže na tubulin. Inhibicí polymerace tubulinu způsobují DM1 i trastuzumab emtansin zastavení buněk ve fázi G2 / M buněčného cyklu, což nakonec vede k buněčné smrti apoptózou. Linker MCC snižuje systémové uvolňování DM1 a zvyšuje jeho koncentraci v cílovém místě. Trastuzumab emtansin je dekonjugován a poté katabolizován proteolýzou v buněčných lysozomech. DM1 je primárně metabolizován CYP3A4 a v menší míře CYP3A5. T0,5 trastuzumabu je přibližně 4 dny. Po opakovaných intravenózních infuzích podávaných v 3týdenním intervalu nedošlo k akumulaci trastuzumab emtansinu. Věk neměl žádný vliv na farmakokinetiku trastuzumab emtansinu.
Dávkování
Intravenózně. Přípravek by měl být předepsán lékařem a podáván pod dohledem zdravotnických pracovníků se zkušenostmi s léčbou onkologických pacientů. Pacienti, kteří dostávají trastuzumab emtansin, by měli mít HER2-pozitivní karcinom - skóre imunohistochemie (IHC) 3+ nebo poměr ≥2 při hybridizaci in situ (ISH). Zkoušky musí být provedeny pomocí diagnostických testů in vitro (IVD) s označením CE. Pokud není k dispozici test CE IVD, měl by být test proveden s jiným validovaným testem. Aby se předešlo lékařským chybám, je důležité zkontrolovat štítek injekční lahvičky a ujistit se, že připravovaným a podávaným léčivým přípravkem je Kadcyla (trastuzumab emtansin) a ne Herceptin (trastuzumab). Doporučená dávka je 3,6 mg / kg. podáván jako intravenózní infuze každé 3 týdny (21denní cyklus). Pacienti by měli být léčeni až do progrese nádoru nebo do dosažení nepřijatelné toxicity. Počáteční dávka by měla být podána jako 90minutová intravenózní infuze. Místo vpichu by mělo být pečlivě sledováno vzhledem k možnosti podkožního průniku léčiva během podávání. Pokud byla předchozí infuze dobře snášena, mohou být následující dávky podány jako 30minutová infuze. Pacienti by měli být sledováni během infuze a po dobu alespoň 30 minut po ní. Rychlost infuze by měla být snížena nebo přerušena, pokud se u pacienta objeví příznaky spojené s infuzí. Trastuzumab emtansin by měl být vysazen v případě život ohrožujících reakcí na infuzi. K okamžitému použití by měly být k dispozici léky na alergické / anafylaktické reakce na infuzi a nouzové vybavení. Pokud dojde k vynechání plánované dávky, měla by být podána co nejdříve; nečekejte až do dalšího cyklu. Dávkovací schéma by mělo být upraveno tak, aby byl zachován 3týdenní dávkovací interval. Další dávka by měla být podána podle doporučení pro dávkování. Léčba symptomatických nežádoucích účinků může zahrnovat periodické vysazení, snížení dávky nebo přerušení léčby. Schéma snížení dávky (počáteční dávka 3,6 mg / kg): první snížení dávky 3 mg / kg; druhé snížení dávky 2,4 mg / kg tělesné hmotnosti; pokud je nutné další snížení dávky, je třeba léčbu ukončit. Pokyny pro úpravu dávky pro zvýšení AST a ALT: Stupeň 2 (> 2,5 až ≤ 5 x ULN) - není nutná žádná úprava dávky; stupeň 3 (> 5 až ≤ 20 x ULN) - přípravek použijte, pokud AST / ALT klesne na stupeň ≤ 2 (> 2,5 až 20 x ULN) - léčbu přerušte. Pravidla úpravy dávky pro hyperbilirubinemii: stupeň 2 (> 1,5 až ≤3 x ULN) - přípravek používejte, když hladina bilirubinu poklesla na stupeň ≤1. (> ULN až 1,5 x ULN), není nutná žádná úprava dávky; Stupeň 3 (> 3 až ≤ 10 x ULN) - přípravek použijte, pokud koncentrace bilirubinu klesne na stupeň ≤ 1 (> ULN na 1,5 x ULN), poté dávku snižte (viz schéma snižování dávky); Stupeň 4 (> 10 x ULN) - ukončení léčby. Pravidla pro úpravu dávky u trombocytopenie: stupeň 3 (koncentrace trombocytů 25 000 až 3) - přípravek použijte, pokud je koncentrace trombocytů ≤ 1. (např. ≥ 75 000 / mm3); není nutná žádná úprava dávky; Stupeň 4 (koncentrace trombocytů 3) - přípravek použijte, když koncentrace trombocytů dosáhne ≤1. (např. ≥ 75 000 / mm3), poté dávku snižte (viz schéma snižování dávky). Zásady úpravy dávky u pacientů s komorovou dysfunkcí: LVEF 45% - pokračujte v léčbě; LVEF 40% až ≤45% se současným snížením ejekční frakce o - přípravek nepoužívejte; Přehodnocení LVEF do 3 týdnů, přerušte léčbu, pokud je LVEF stále ≥ 10 procentních bodů od výchozí hodnoty; symptomatický CHF - ukončete léčbu. Děti a dospívající: Bezpečnost a účinnost u dětí a dospívajících do 18 let nebyla stanovena, protože u této populace není známo, že diseminovaný karcinom prsu (MBC). Zvláštní skupiny pacientů. Léčba by měla být dočasně přerušena u pacientů, u kterých se vyvine periferní neuropatie 3. nebo 4. stupně, dokud nedosáhne stupně ≤ 2; při opětovném zahájení léčby je možné zvážit snížení dávky, jak je uvedeno v plánu snižování dávky. U pacientů ve věku ≥ 65 let není nutná žádná úprava dávky; nejsou k dispozici dostatečné údaje k určení bezpečnosti a účinnosti léčby u pacientů ve věku ≥ 75 let. U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin není nutná žádná úprava počáteční dávky; Potenciální potřebu úpravy dávky u pacientů se závažnou renální nedostatečností nelze určit, a proto by tito pacienti měli být pečlivě sledováni. U pacientů s mírnou nebo středně těžkou poruchou funkce jater není nutná žádná úprava počáteční dávky. Trastuzumab emtansin nebyl studován u pacientů s těžkou poruchou funkce jater; při léčbě pacientů s poruchou funkce jater je nutná opatrnost kvůli známé hepatotoxicitě pozorované během léčby. Způsob dávání. Přípravek by měl být rozpuštěn a naředěn zdravotnickým personálem a podáván jako intravenózní infuze. Nesmí být podán jako bolus nebo jako rychlá injekce.
Indikace
Monoterapie dospělých pacientů s HER2-pozitivním, neoperovatelným lokálně pokročilým nebo metastazujícím karcinomem prsu, dříve léčených trastuzumabem a taxanem, v kombinaci nebo samostatně. Pacienti, kteří podstoupili předchozí léčbu lokálně pokročilého nebo generalizovaného onemocnění nebo u nichž došlo k relapsu během nebo do 6 měsíců od ukončení adjuvantní léčby.
Kontraindikace
Přecitlivělost na účinnou látku nebo jiné složky přípravku.
Opatření
V klinických studiích s trastuzumabem emtansinem byly hlášeny případy intersticiálního plicního onemocnění (ILD), včetně pneumonie; některé byly spojeny s akutním respiračním selháním nebo byly smrtelné. U pacientů s diagnostikovanou ILD nebo pneumonií se doporučuje přerušení léčby přípravkem. U pacientů s dušností v klidu spojenou s komplikacemi pokročilého karcinomu a přidruženými chorobami může být zvýšené riziko vzniku plicních komplikací. Během léčby přípravkem byla pozorována hepatotoxicita a závažné poruchy jater a žlučových cest, včetně nodulární regenerativní hypertrofie (NRH) jater a úmrtí v důsledku poškození jater vyvolaného léky; mohly být také ovlivněny komorbidity a / nebo souběžné léky, o nichž je známo, že mají hepatotoxický potenciál. Funkce jater by měla být sledována před zahájením léčby a před následným podáním dávky. Pacienti s výchozím zvýšením ALT (např. Spojeným s jaterními metastázami) jsou náchylní k rozvoji selhání jater a je u nich vyšší riziko jaterní toxicity stupně 3 nebo zvýšené laboratorní hladiny jater. Případy NRH byly diagnostikovány biopsiemi jater. O vývoji NRH je třeba uvažovat u všech pacientů s klinickými příznaky portální hypertenze a / nebo obrazem podobným cirhóze pozorovaným na CT skenu jater, avšak s normálními hladinami sérových transamináz a bez dalších známek cirhózy. Pokud je diagnostikován NRH, měla by být léčba přípravkem přerušena. Nebylo studováno u pacientů se sérovými transaminázami> 2,5násobek ULN (horní hranice normálu) nebo s celkovým bilirubinem> 1,5násobek ULN před zahájením léčby. V případě aktivity sérových transamináz> 3 x ULN a současně celkové koncentrace bilirubinu> 2 x ULN by měla být léčba ukončena. Při léčbě pacientů s poruchou funkce jater je nutná opatrnost. Během léčby existuje zvýšené riziko dysfunkce levé komory. U pacientů léčených trastuzumabem emtansinem bylo pozorováno snížení ejekční frakce levé komory (LVEF) po 50 letech) s počáteční nízkou hodnotou LVEF (25 kg / m2). Před zahájením léčby a v pravidelných intervalech (např. Každé 3 měsíce) je třeba provést standardní testy srdeční funkce (echokardiogram nebo radionuklidová angiografie s více branami). Výchozí hodnota LVEF pacientů byla ve většině klinických studií ≥ 50%. Ze studie byli vyloučeni pacienti s městnavým srdečním selháním, těžkými arytmiemi vyžadujícími léčbu, anamnézou infarktu myokardu nebo nestabilní anginou pectoris během 6 měsíců před randomizací nebo s klidovou dušností v důsledku pokročilého neoplastického onemocnění. V případě poruch levé komory je třeba odložit podání další dávky nebo ukončit léčbu. Účinek léčby trastuzumabem emtansinem u pacientů, kteří ukončili léčbu trastuzumabem kvůli reakci na infuzi, nebyl studován; léčba touto drogou se u této skupiny pacientů nedoporučuje. Léčba by měla být přerušena u pacientů, u kterých se rozvine závažná reakce spojená s infuzí, dokud příznaky nevymizí. Opětovné zahájení léčby by mělo být zváženo na základě klinického posouzení závažnosti reakce. Léčba by měla být přerušena v případě život ohrožující reakce na infuzi. Účinek léčby trastuzumabem emtansinem u pacientů, kteří přerušili léčbu trastuzumabem z důvodu přecitlivělosti, nebyl studován; Použití trastuzumab emtansinu u těchto pacientů se nedoporučuje. Pacienti by měli být sledováni z hlediska přecitlivělosti / alergických reakcí, jejichž příznaky mohou být stejné jako reakce související s infuzí; Byly pozorovány závažné anafylaktické reakce. Pokud dojde k přecitlivělosti (se zvýšenými reakcemi během následujících infuzí), měla by být léčba trastuzumabem emtansinem přerušena.Vzhledem k riziku hemoragických příhod (včetně CNS, respiračního a gastrointestinálního krvácení) je nutná opatrnost a při současném podávání s antikoagulancii nebo antiagregačními léky je třeba zvážit další sledování. Před podáním každé dávky trastuzumabu emtansinu se doporučuje sledovat počet krevních destiček. Pacienti s trombocytopenií (≤ 100 000 / mm3) a pacienti léčení antikoagulancii (např. Warfarin, heparin, nízkomolekulární hepariny) by měli být během léčby trastuzumabem emtansinem pečlivě sledováni. Trastuzumab emtansin nebyl studován u pacientů s počtem trombocytů ≤ 100 000 / mm3 před zahájením léčby. V případě zhoršení trombocytopenie na stupeň 3 nebo vyšší (3) by se přípravek neměl používat, dokud toxicita neklesne na stupeň 1 (≥ 75 000 / mm3). V klinických studiích s trastuzumabem emtansinem se vyskytla periferní neuropatie 1. stupně, zejména senzorická. Pacienti s periferní neuropatií stupně ≥ 3 byli z účasti v klinických studiích vyloučeni. Léčba pacientů s periferní neuropatií stupně 3 nebo 4 by měla být pravidelně přerušována, dokud se komplikace nesníží na stupeň ≤2. Pacienti by měli být pravidelně sledováni kvůli známkám neurotoxicity. Aby se zlepšila sledovatelnost biologických přípravků, měl by být obchodní název podávaného léčiva jasně zapsán (nebo uveden) v dokumentaci pacienta. Bezpečnost a účinnost léku u dětí a dospívajících do 18 let nebyla stanovena, protože u této populace nebyl zjištěn rozšířený karcinom prsu. Nesmí být podán jako bolus nebo jako rychlá injekce.
Nežádoucí aktivita
Velmi časté: infekce močových cest, trombocytopenie, anémie, hypokalémie, nespavost, periferní neuropatie, bolest hlavy, krvácení, epistaxe, kašel, dušnost, stomatitida, průjem, zvracení, nevolnost, zácpa, sucho v ústech, bolest břicha vyrážka, muskuloskeletální bolest, artralgie, myalgie, únava, horečka, astenie, zimnice, zvýšení transamináz. Časté: neutropenie, leukopenie, přecitlivělost, závratě, dysgeuzie, poruchy paměti, syndrom suchého oka, konjunktivitida, poruchy zraku, zvýšené slzení, dysfunkce levé komory, hypertenze, dyspepsie, krvácení z dásní, svědění kůže, alopecie, onemocnění nehtů palmoplantární erytrodysestézie, kopřivka, periferní edém, zvýšení alkalické fosfatázy, reakce související s infuzí (zarudnutí kůže, zimnice, horečka, dušnost, nízký krevní tlak, sípání, bronchospazmus, tachykardie). Méně časté: pneumonie (ILD), hepatotoxicita, selhání jater, nodulární regenerační hyperplazie, portální hypertenze, extravazace v místě vpichu (erytém, citlivost, podráždění kůže, bolest nebo otok). Kromě toho byla pozorována hyperbilirubinemie. V klinických studiích došlo během léčby trastuzumabem emtansinem ke zvýšení sérových transamináz (stupeň 1-4) a bylo obvykle přechodné. Zvýšení transamináz bylo nejčastěji přechodné a vyvrcholilo 8. den po podání. Poté toxicita poklesla na stupeň 1 nebo odezněla před dalším cyklem. Došlo také k kumulativnímu účinku (procento pacientů se zvýšením ALT / AST stupně 1-2 se v následujících cyklech zvýšilo). U pacientů se zvýšenými hladinami transamináz došlo u většiny pacientů ke snížení nebo zotavení toxicity 1. stupně do 30 dnů od poslední dávky trastuzumabu emtansinu. Dysfunkce levé komory byla zjištěna u 2,2% pacientů účastnících se klinických studií. Ve většině případů bylo snížení LVEF stupně 1 nebo 2 asymptomatické. Toxicita stupně 3 nebo 4 byla pozorována u 0,4% pacientů, obvykle během počátečních cyklů léčby (1–2). Závažné epizody hemoragických příhod (stupeň ≥3) byly hlášeny u 2,2% všech pacientů. Trombocytopenie se vyskytla u 24,9% pacientů v klinických studiích a byla nejčastějším nežádoucím účinkem vedoucím k přerušení léčby. V klinických studiích byl výskyt a závažnost trombocytopenie vyšší u asijských pacientů. Někteří pacienti, u kterých se tyto komplikace vyvinuly, byli také léčeni antikoagulací. Vyskytly se epizody smrtelného krvácení a závažné komplikace krvácení, včetně krvácení do CNS, 5,3% pacientů mělo pozitivní test na protilátky proti trastuzumabu emtansinu.
Těhotenství a kojení
Užívání trastuzumab emtansinu u těhotných žen se nedoporučuje. Před těhotenstvím by ženy měly být informovány o možnosti poškození plodu. Pacientky, které otěhotní, by však měly okamžitě kontaktovat lékaře. Pokud je těhotná žena léčena trastuzumabem emtansinem, doporučuje se pečlivé sledování multidisciplinárním týmem. Trastuzumab může způsobit poškození plodu nebo smrt, pokud je podáván těhotné ženě. U dětí těhotných žen léčených přípravkem byly hlášeny případy oligohydramnionu, z nichž některé vedly k fatální hypoplázii plic. DM1, cytotoxická složka trastuzumab emtansinu, může být teratogenní a potenciálně embryotoxická. Ženy by měly před zahájením léčby trastuzumabem emtansinem ukončit kojení. Pacientky mohou začít kojit 7 měsíců po ukončení léčby. Ženy ve fertilním věku by měly během léčby tímto přípravkem a po dobu 7 měsíců po poslední dávce trastuzumab emtansinu používat účinnou antikoncepci. Efektivní metody antikoncepce by měli používat také muži nebo jejich partnerky.
Komentáře
Pacienti s reakcemi souvisejícími s infuzí by neměli řídit nebo obsluhovat stroje, dokud tyto příznaky nevymizí. Informace připravené na základě SPC 13. července 2017 Aktuální SmPC je k dispozici na www.roche.pl.
Interakce
Výsledky in vitro studií metabolismu v lidských jaterních mikrozomech naznačují, že DM1 je metabolizován primárně enzymem CYP3A4 a v menší míře CYP3A5. Je třeba se vyvarovat současného užívání silných inhibitorů CYP3A4 (např. Ketokonazolu, itrakonazolu, klarithromycinu, atazanaviru, indinaviru, nefazodonu, nelfinaviru, ritonaviru, sachinaviru, telithromycinu a vorikonazolu), protože to může zvýšit hladinu a toxicitu DM1. Měla by se zvážit alternativní formulace, která neinhibuje nebo jen mírně inhibuje CYP3A4. Pokud nelze zabránit současnému užívání silných inhibitorů CYP3A4, a pokud je to možné, zvažte odložení podávání trastuzumab emtansinu, dokud nebudou inhibitory CYP3A4 odstraněny z oběhu (poločasy přibližně 3 inhibitorů). Pokud se však současně užívá silný inhibitor CYP3A4 a léčbu trastuzumabem emtansinem nelze odložit, měl by být pacient v takových případech pečlivě sledován z hlediska nežádoucích účinků.
Přípravek obsahuje látku: Trastuzumab emtansin
Úhrada léku: NE